Općenito
CHARMM (Chemistry at HARvard Macromolecular Mechanics) softver je za simulacije molekulske dinamike sa širokim spektrom aplikacija na sustave s velikim brojem čestica. Podržava tehnike različitih veličina skala kao i analitičkih alata. Primarno je osmišljen za proučavanje bioloških sustava poput peptida, proteina, prostetičkih skupina, liganda, nukleinskih kiselina, lipida i ugljikohidrata, a prilagođen je za rad s kristalima, otopinama te membranama. Također, koristi se i kod dizajna anorganskih materijala.
CHARMM je na računalnom klasteru Isabella kompajliran s Intelovim C/C++ i Fortran kompajlerima te koristi Open MPI knjižnice. Podržava serijsko i paralelno izvođenje poslova, odnosno izvođenje na jednojezgrenim ili višejezgrenim procesorima.
Moduli
Moduli koji dopremaju CHARMM u vašu okolinu definirani su u tablici niže:
| Verzija | Modul |
|---|
| 47b1 | charmm/47b1 |
Primjer korištenja
Primjer serijskog korištenja
#!/bin/bash
#$ -N charmm
#$ -cwd
module load charmm/47b1
charmm < input.inp > output.out
Primjer paralelnog korištenja
#!/bin/bash
#$ -N charmm
#$ -cwd
#$ -pe *mpi 4
module load charmm/47b1
mpirun -np ${NSLOTS} charmm -i input.inp -o output.out
Praktični primjer
Simulacija molekulske dinamike sustava sačinjenog od 32 molekule vode
Simulacija molekulske dinamike sustava koji se sastoji od 32 molekule vode koristeći uobičajeni način izvršavanja poslova putem skripti. Sve naredbe mogu se podnijeti i zasebno koristeći tzv. interaktivni model.
Ulazna (engl. input) datoteka sadrži nekoliko cjelina:
- RTF (engl. residue topology file) dio koji opisuje molekulu vode (rezidua),
- dio koji opisuje parametre rezidue,
- dio koji generira određeni broj opisane rezidue,
- dio koji smješta generirane rezidue unutar koordinatnog sustava,
- dio koji postavlja periodične uvjete simulacije i
- dio koji zadaje uvjete simulacije molekulske dinamike te dio koji ispisuje rezultat.
RTF dio sadrži tipove atome i njihove mase, proučavanu reziduu i točan broj njezinih atoma i imena, međuatomske veze i njihove kutove te terminalni dio molekule. Potom se upisuju parametri polja sila potrebni za opis duljina veza i kuteva rezidue od interesa poput konstanti sila, van der Wallsovih radijusa i sl. Uvjeti simulacije molekulske dinamike najvarijabilniji je i najkompleksniji je dio same skripte te direktno ovisi o samoj prirodi simuliranog sustava. Dio koji postavlja periodične uvjete esencijalni je dio metode molekulske dinamike te služi kako bi proučavani sustav bio što realističnija reprezentacija prirodnog; centralni sustav okružuje se kopijama samog sebe kako bi se, između ostalog, očuvale interakcije čestica sustava. Zahtijeva opis simulacijske kutije koja je u ovom primjeru kubičnog oblika.
* CHARMM input script to run an MD simulation on a
* system of 32 water molecules
BOMLEV -3
READ RTF CARD
* This is the water residue
*
47 1
MASS 1 H 1.0 ! Water - Hydrogen Atom
MASS 2 O 16.0 ! Water - Oxygen Atom
RESIDUE H2O 0.0 ! Water Model and its charge
ATOM OH2 O -0.82 ! Oxygen atom and charge
ATOM H1 H 0.41 ! Hydrogen atom and charge
ATOM H2 H 0.41 ! Hydrogen atom and charge
BOND OH2 H1 OH2 H2 ! Define atom-atom connections
THETA H1 OH2 H2 ! Define dihedral angle
PATCH FIRST NONE LAST NONE ! Do not patch the termini
END
! ===================================================================
READ PARAMETERS CARD
* Bond and angle constraints for water
*
BOND
H O 450.0 0.9572 ! Force constant of the H-O bond
THETA
H O H 55.0 104.52 ! Equilibrium angle in degrees
NONBONDED
O 0.0 -0.1521 1.7682 ! Well-depth and van der Waals radius
H 0.0 -0.04598 0.2245 ! Well-depth and van der Waals radium
END
! ===================================================================
READ SEQUENCE H2O 32
GENERATE WAT SETUP
READ COORDINATES CARD FREE
* Oxygen Coordinates
*
32
1 1 H2O OH2 -4.5 -4.5 -4.5
1 2 H2O OH2 -1.5 -4.5 -4.5
1 3 H2O OH2 -4.5 -1.5 -4.5
1 4 H2O OH2 -4.5 -4.5 -1.5
1 5 H2O OH2 -1.5 -1.5 -4.5
1 6 H2O OH2 -1.5 -4.5 -1.5
1 7 H2O OH2 -4.5 -1.5 -1.5
1 8 H2O OH2 -1.5 -1.5 -1.5
1 9 H2O OH2 -4.5 -4.5 1.5
1 10 H2O OH2 -1.5 -4.5 1.5
1 11 H2O OH2 -4.5 -1.5 1.5
1 12 H2O OH2 -4.5 -4.5 4.5
1 13 H2O OH2 -1.5 -1.5 1.5
1 14 H2O OH2 -1.5 -4.5 4.5
1 15 H2O OH2 -4.5 -1.5 4.5
1 16 H2O OH2 -1.5 -1.5 4.5
1 17 H2O OH2 -4.5 1.5 -4.5
1 18 H2O OH2 -1.5 1.5 -4.5
1 19 H2O OH2 -4.5 4.5 -4.5
1 20 H2O OH2 -4.5 1.5 -1.5
1 21 H2O OH2 -1.5 4.5 -4.5
1 22 H2O OH2 -1.5 1.5 -1.5
1 23 H2O OH2 -4.5 4.5 -1.5
1 24 H2O OH2 -1.5 4.5 -1.5
1 25 H2O OH2 -4.5 1.5 1.5
1 26 H2O OH2 -1.5 1.5 1.5
1 27 H2O OH2 -4.5 4.5 1.5
1 28 H2O OH2 -4.5 1.5 4.5
1 29 H2O OH2 -1.5 4.5 1.5
1 30 H2O OH2 -1.5 1.5 4.5
1 31 H2O OH2 -4.5 4.5 4.5
1 32 H2O OH2 -1.5 4.5 4.5
! format above is (segment #) (residue #) (residue name) (atom type)
! (x coor) (y coor) (z coor)
HBUILD
PRINT COORDINATES
! The psf file contains all the information on the makeup of the system
OPEN UNIT 1 WRITE CARD NAME water.psf
WRITE PSF CARD UNIT 1
* PSF for 32 water molecule system
*
! The CRD file will contain the coordinates of all the atoms in the system
! written in CHARMM format
OPEN UNIT 1 WRITE CARD NAME water.crd
WRITE COORDINATE CARD UNIT 1
* Initial coordinates for starting point of simulation
*
! The PDB file will contain the coordinates of all the atoms in the system
! written in Protein Data Bank format
OPEN UNIT 1 WRITE CARD NAME water.pdb
WRITE COORDINATES PDB UNIT 1
* Initial coordinates for starting point of simulation
*
CRYSTAL DEFINE CUBIC 13.0 13.0 13.0 90.0 90.0 90.0
CRYSTAL BUILD NOPERATIONS 0
OPEN UNIT 1 WRITE CARD NAME cubic.cry
WRITE CRYSTAL CARD UNIT 1
* PBC for simple cubic geometry
*
SHAKE BONH PARAMETERS SELECT ALL END ! Constrain all bonds to H atoms
! to their parameter values
OPEN UNIT 11 WRITE FILE NAME water.dcd
DYNAMICS STRT -
NSTEP 20000 TIMESTEP 0.001 -
NPRINT 100 IPRFRQ 1000 NTRFRQ 5000 -
NSAVC 100 IUNCRD 11 IUNWRI 1 -
FIRSTT 100.0 FINALT 300.0 TEMINC 50.0 IHTFRQ 500 -
IASORS 1 IASVEL 1 ISCVEL 0 IEQFRQ 1000 ICHECW 0 -
TWINDH 10.0 TWINDL -10.0 -
INBFRQ 25 IMGFRQ 25 CUTNB 12.0 CUTIM 12.0 CTOFNB 10.0 -
CTONNB 8.0 SHIFT VSWITCH
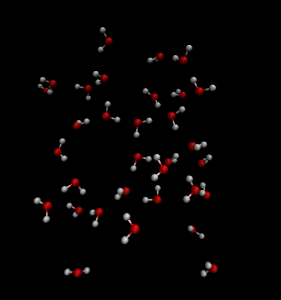
Slika 1 Posljednja struktura sustava koji sadrži 32 molekule vode nastala dinamičkom evolucijom u vremenu.